OXtal: An All-Atom Diffusion Model for Organic Crystal Structure Prediction
Dec 13, 2025·,,,,,,,,,,
Emily Jin*
Andrei Cristian Nica*
Mikhail Galkin
Jarrid Rector-Brooks
Kin Long Kelvin Lee
Santiago Miret
Frances H. Arnold
Michael Bronstein
Joey Bose
Alexander Tong
Cheng-Hao Liu
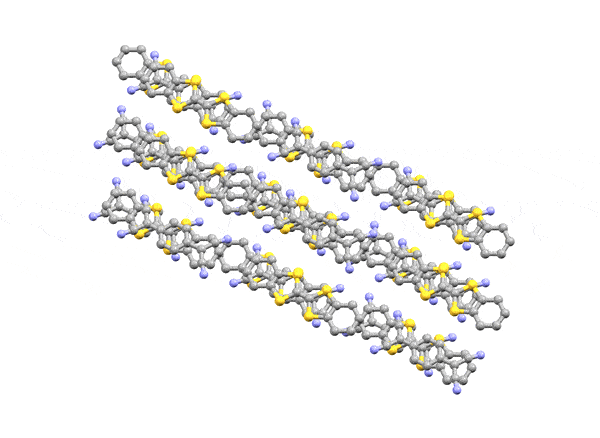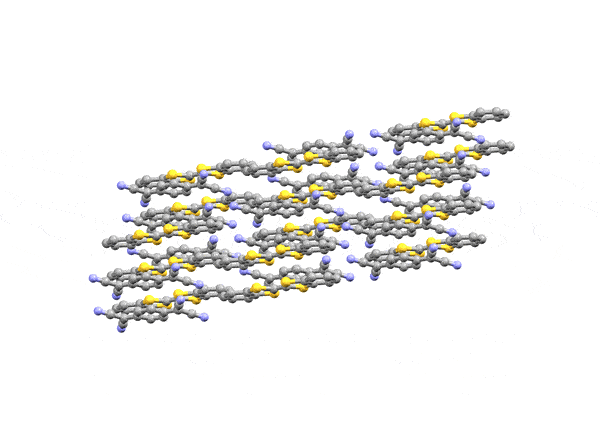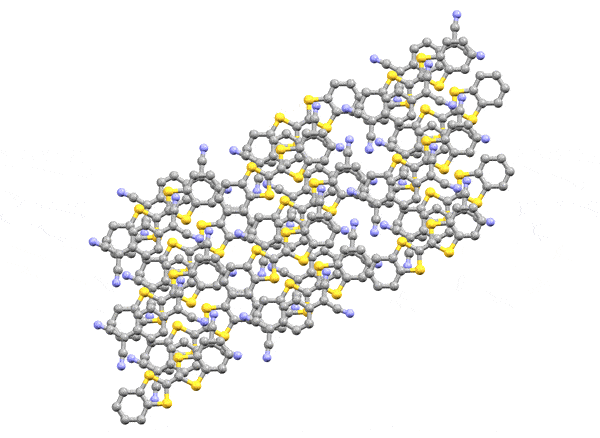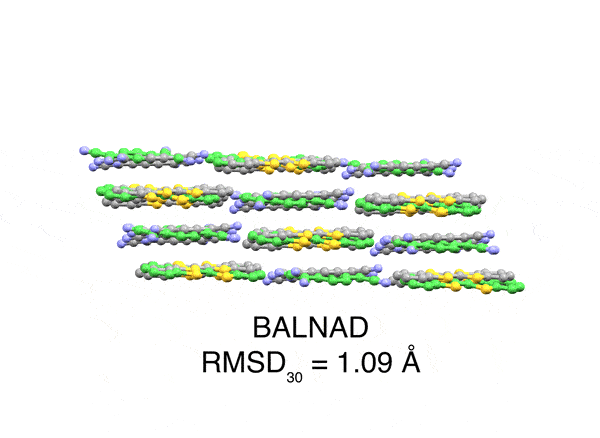
Abstract
Accurately predicting experimentally-realizable 3D molecular crystal structures from their 2D chemical graphs is a long-standing open challenge in computational chemistry called crystal structure prediction (CSP). Efficiently solving this problem has implications ranging from pharmaceuticals to organic semiconductors, as crystal packing directly governs the physical and chemical properties of organic solids. In this paper, we introduce OXtal, a large-scale 100M parameter all-atom diffusion model that directly learns the conditional joint distribution over intramolecular conformations and periodic packing. To efficiently scale OXtal, we abandon explicit equivariant architectures imposing inductive bias arising from crystal symmetries in favor of data augmentation strategies. We further propose a novel crystallization-inspired lattice-free training scheme, Stoichiometric Stochastic Shell Sampling ($S^4$), that efficiently captures long-range interactions while sidestepping explicit lattice parametrization – thus enabling more scalable architectural choices at all-atom resolution. By leveraging a large dataset of 600K experimentally validated crystal structures (including rigid and flexible molecules, co-crystals, and solvates), OXtal achieves orders-of-magnitude improvements over prior ab initio machine learning CSP methods, while remaining orders of magnitude cheaper than traditional quantum-chemical approaches. Specifically, OXtal recovers experimental structures with conformer RMSD${}_1 < 0.5$ Å and attains over 80% packing similarity rate, demonstrating its ability to model both thermodynamic and kinetic regularities of molecular crystallization.
Type
Publication
arXiv